Maximum-likelihood estimation of recent shared ancestry (ERSA)
[tpsingle id="26"]ERSA (Estimation of Recent Shared Ancestry) estimates recent shared ancestry between pairs of individuals based on the number and lengths of chromosomal segments that they share identically-by-descent through common ancestors (IBD segments). Prior to ERSA, established methods were capable of accurately estimating kinship for first-degree through third-degree relatives. ERSA is accurate to within one degree of relationship for 97% of first-degree through fifth-degree relatives and 80% of sixth-degree and seventh-degree relatives, greatly expanding the range of relationships that can be estimated from genetic data.
Relationship Estimation from Whole-Genome Sequence Data (ERSA 2.0)
[tpsingle id="35"]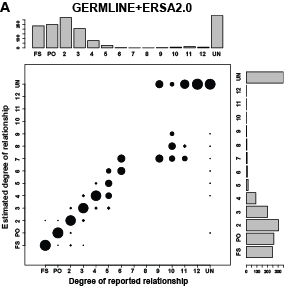
Licensing and Download
Software implementing the ERSA algorithm is now available. Register to download the most recent version (2.0) here.
Release Notes
Version 2.1
- Added a new parameter: model_output_file. When this parameter is specified, a new file is created that reports the likelihood for every possible pairwise relationship.
- Fixed an issue in which pairs of individuals that shared no segments IBD of the minimum length or greater would be absent in the output file. Now, all possible pairs of individuals are included in the output file, regardless of whether they share any segments IBD. Note that individuals sharing no IBD segments will always be reported as unrelated.
All Versions
- version 2.1 (current)
- version 1.0